








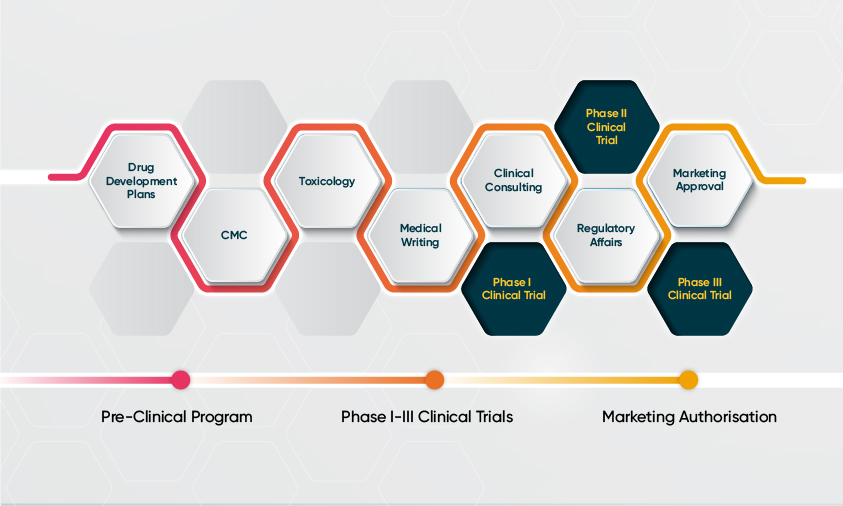
Drug Development Strategies
The right development strategy is critical for commercial success. With biotech companies investing in advanced therapies and international trials, there is an increased focus on regulatory planning, including the design of the clinical protocols, nonclinical testing plans and manufacturing processes. Novotech provides first-class regulatory services to help expedite drug development within the complex and dynamic research environment.
Core Regulatory Services
- Regulatory gap assessment and strategy
- Design and review of nonclinical toxicology studies
- Writing Investigator Brochures (IBs)
- Global submissions including IND, DMF, NDA, BLA and combination products
- Meetings and briefing packages including Type A, B and C meetings, scientific advice
- Fast track and orphan designations
- USA agent services
- Institutional Biosafety Committee and GMO medicine applications
Core Medical Services
- Medical writing
- Clinical trial design
- Medical monitoring
- Pharmacovigilance
From inception, through development and to approval, Novotech Drug Development Consulting is with you at every step.